ADN
Problème à résoudre
L'ADN, porteur de l'information génétique des êtres vivants, est utilisé dans la justice pénale depuis des décennies. Mais comment, exactement, fonctionne le profilage ADN ? Étant donné une séquence d'ADN, comment les enquêteurs médico-légaux peuvent-ils identifier à qui elle appartient ?
Dans un fichier nommé dna.py dans un dossier nommé dna, implémentez un programme qui identifie à qui appartient une séquence d'ADN.
Démo
Code de distribution
Pour ce problème, vous allez étendre les fonctionnalités du code fourni par le personnel de CS50.
Connectez-vous à cs50.dev, cliquez sur votre fenêtre de terminal et exécutez cd seul. Vous devriez constater que l'invite de votre fenêtre de terminal ressemble à ce qui suit :
$
Ensuite, exécutez :
wget https://cdn.cs50.net/2023/fall/psets/6/dna.zip
afin de télécharger un ZIP nommé dna.zip dans votre codespace.
Ensuite, exécutez :
unzip dna.zip
pour créer un dossier nommé dna. Vous n'avez plus besoin du fichier ZIP, vous pouvez donc exécuter :
rm dna.zip
et répondre par « y » suivi d'Entrée à l'invite pour supprimer le fichier ZIP que vous avez téléchargé.
Maintenant, tapez :
cd dna
suivi d'Entrée pour vous déplacer dans (c'est-à-dire ouvrir) ce répertoire. Votre invite devrait maintenant ressembler à ceci :
dna/ $
Exécutez ls seul, et vous devriez voir quelques fichiers et dossiers :
databases/ dna.py sequences/
Si vous rencontrez des problèmes, suivez à nouveau ces mêmes étapes et voyez si vous pouvez déterminer où vous vous êtes trompé !
Contexte
L'ADN n'est en réalité qu'une séquence de molécules appelées nucléotides, disposées selon une forme particulière (une double hélice). Chaque cellule humaine contient des milliards de nucléotides disposés en séquence. Chaque nucléotide d'ADN contient l'une des quatre bases différentes : l'adénine (A), la cytosine (C), la guanine (G) ou la thymine (T). Certaines parties de cette séquence (c'est-à-dire le génome) sont identiques, ou du moins très similaires, chez presque tous les humains, mais d'autres parties de la séquence ont une plus grande diversité génétique et varient donc davantage au sein de la population.
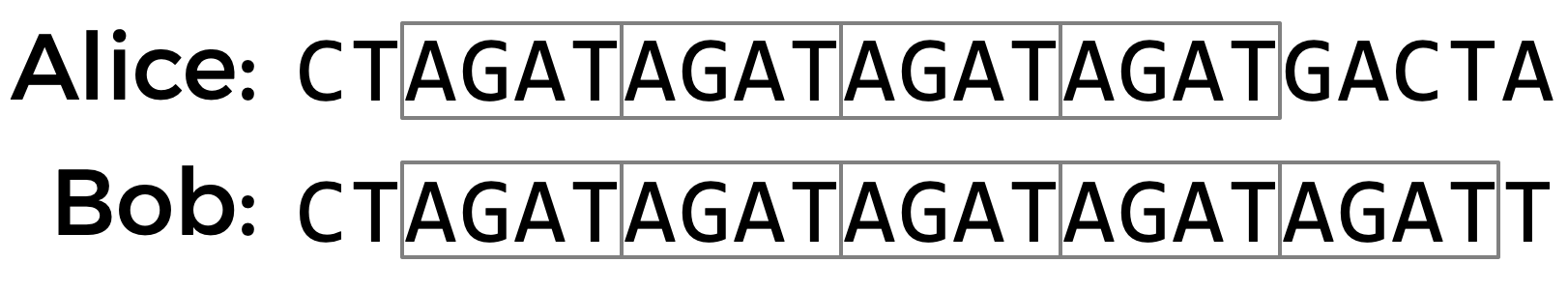
Un endroit où l'ADN a tendance à avoir une grande diversité génétique se trouve dans les répétitions en tandem courtes (STR). Une STR est une courte séquence de bases ADN qui tend à se répéter consécutivement de nombreuses fois à des endroits spécifiques dans l'ADN d'une personne. Le nombre de répétitions d'une STR particulière varie beaucoup d'un individu à l'autre. Dans les échantillons d'ADN ci-dessous, par exemple, Alice a la STR AGAT répétée quatre fois dans son ADN, tandis que Bob a la même STR répétée cinq fois.
L'utilisation de plusieurs STR, plutôt qu'une seule, peut améliorer la précision du profilage ADN. Si la probabilité que deux personnes aient le même nombre de répétitions pour une seule STR est de 5 %, et que l'analyste examine 10 STR différentes, alors la probabilité que deux échantillons d'ADN correspondent par pur hasard est d'environ 1 sur 1 quadrillion (en supposant que toutes les STR sont indépendantes les unes des autres). Ainsi, si deux échantillons d'ADN correspondent au nombre de répétitions pour chacune des STR, l'analyste peut être assez certain qu'ils proviennent de la même personne. CODIS, la base de données ADN du FBI, utilise 20 STR différentes dans le cadre de son processus de profilage ADN.
À quoi pourrait ressembler une telle base de données ADN ? Eh bien, dans sa forme la plus simple, vous pouvez imaginer formater une base de données ADN sous forme de fichier CSV, dans lequel chaque ligne correspond à un individu et chaque colonne correspond à une STR particulière.
nom, AGAT, AATG, TATC
Alice, 28, 42, 14
Bob, 17, 22, 19
Charlie, 36, 18, 25
Les données du fichier ci-dessus suggèrent qu'Alice a la séquence AGAT répétée 28 fois consécutivement quelque part dans son ADN, la séquence AATG répétée 42 fois et TATC répétée 14 fois. Bob, quant à lui, a ces trois mêmes STR répétées 17 fois, 22 fois et 19 fois, respectivement. Et Charlie a ces trois mêmes STR répétées 36, 18 et 25 fois, respectivement.
Alors, étant donné une séquence d'ADN, comment pouvez-vous identifier à qui elle appartient ? Eh bien, imaginez que vous ayez parcouru la séquence d'ADN à la recherche de la plus longue séquence consécutive d'AGAT répétés et que vous ayez trouvé que la plus longue séquence faisait 17 répétitions de long. Si vous trouviez ensuite que la plus longue séquence d'AATG faisait 22 répétitions de long et que la plus longue séquence de TATC avait 19 répétitions de long, cela fournirait de très bonnes preuves que l'ADN était celui de Bob. Bien sûr, il est également possible qu'une fois que vous ayez pris les comptes pour chacune des STR, il ne corresponde à personne dans votre base de données ADN, auquel cas vous n'avez aucune correspondance.
Dans la pratique, comme les analystes savent sur quel chromosome et à quel endroit de l'ADN une STR se trouvera, ils peuvent localiser leur recherche sur une section étroite d'ADN. Mais nous ignorerons ce détail pour ce problème.
Votre tâche consiste à écrire un programme qui prendra une séquence d'ADN et un fichier CSV contenant les comptes de STR pour une liste d'individus, puis sortira à qui l'ADN (très probablement) appartient.
Spécifications
- Le programme doit exiger comme premier argument de ligne de commande le nom d'un fichier CSV contenant les comptes de STR pour une liste d'individus et doit exiger comme deuxième argument de la ligne de commande le nom d'un fichier texte contenant la séquence ADN à identifier.
- Si votre programme est exécuté avec un nombre incorrect d'arguments de ligne de commande, votre programme doit imprimer un message d'erreur de votre choix (avec
print). Si le nombre correct d'arguments est fourni, vous pouvez supposer que le premier argument est bien le nom de fichier d'un fichier CSV valide et que le deuxième argument est le nom de fichier d'un fichier texte valide. - Votre programme doit ouvrir le fichier CSV et lire son contenu en mémoire.
- Vous pouvez supposer que la première ligne du fichier CSV sera les noms des colonnes. La première colonne sera le mot
nomet les colonnes restantes seront les séquences STR elles-mêmes. - Votre programme doit ouvrir la séquence d'ADN et lire son contenu en mémoire.
- Pour chacune des STR (à partir de la première ligne du fichier CSV), votre programme doit calculer la plus longue série de répétitions consécutives de la STR dans la séquence d'ADN à identifier. Notez que nous avons défini une fonction d'assistance pour vous,
longest_match, qui fera exactement cela ! - Si les comptes de STR correspondent exactement à l'un des individus du fichier CSV, votre programme doit imprimer le nom de l'individu correspondant. - Vous pouvez supposer que les comptes de STR ne correspondront pas à plus d'un individu.
- Si les comptes de STR ne correspondent pas exactement à l'un des individus du fichier CSV, votre programme doit imprimer
Aucune correspondance.
Conseils
-
Le module
csvpeut vous être utile pour lire les fichiers CSV en mémoire.csv.DictReaderpeut être particulièrement utile.-
Par exemple, si un fichier comme
foo.csvcomporte une ligne d'en-tête dans laquelle chaque chaîne de caractères correspond au nom d'un champ, voici comment vous pouvez imprimer cesfieldnamesen tant quelist: import csvwith open("foo.csv") as file: reader = csv.DictReader(file) print(reader.fieldnames) -
Et voici comment lire toutes les (autres) lignes d'un CSV dans une
list, dans laquelle chaque élément est undictqui représente cette ligne : import csvrows = [] with open("foo.csv") as file: reader = csv.DictReader(file) for row in reader: rows.append(row)
-
-
Les fonctions
openetreadpeuvent également se révéler utiles pour lire des fichiers texte en mémoire. - Réfléchissez aux structures de données qui pourraient vous être utiles pour suivre les informations dans votre programme. Une
listou undictpeut s'avérer utile. - N'oubliez pas que nous avons défini une fonction (
longest_match) qui, lorsqu'on lui fournit une séquence ADN et une STR en entrée, renvoie le nombre maximum de fois que la STR se répète. Vous pouvez ensuite utiliser cette fonction dans d'autres parties de votre programme !
Procédure pas à pas
Comment tester
Bien que check50 soit disponible pour ce problème, nous vous encourageons à tester votre code par vous-même pour chacun des tests suivants.
- Exécutez votre programme avec
python dna.py databases/small.csv sequences/1.txt. Votre programme doit afficherBob. - Exécutez votre programme avec
python dna.py databases/small.csv sequences/2.txt. Votre programme doit afficherNo match. - Exécutez votre programme avec
python dna.py databases/small.csv sequences/3.txt. Votre programme doit afficherNo match. - Exécutez votre programme avec
python dna.py databases/small.csv sequences/4.txt. Votre programme doit afficherAlice. - Exécutez votre programme avec
python dna.py databases/large.csv sequences/5.txt. Votre programme doit afficherLavender. - Exécutez votre programme avec
python dna.py databases/large.csv sequences/6.txt. Votre programme doit afficherLuna. - Exécutez votre programme avec
python dna.py databases/large.csv sequences/7.txt. Votre programme doit afficherRon. - Exécutez votre programme avec
python dna.py databases/large.csv sequences/8.txt. Votre programme doit afficherGinny. - Exécutez votre programme avec
python dna.py databases/large.csv sequences/9.txt. Votre programme doit afficherDraco. - Exécutez votre programme avec
python dna.py databases/large.csv sequences/10.txt. Votre programme doit afficherAlbus. - Exécutez votre programme avec
python dna.py databases/large.csv sequences/11.txt. Votre programme doit afficherHermione. - Exécutez votre programme avec
python dna.py databases/large.csv sequences/12.txt. Votre programme doit afficherLily. - Exécutez votre programme avec
python dna.py databases/large.csv sequences/13.txt. Votre programme doit afficherNo match. - Exécutez votre programme avec
python dna.py databases/large.csv sequences/14.txt. Votre programme doit afficherSeverus. - Exécutez votre programme avec
python dna.py databases/large.csv sequences/15.txt. Votre programme doit afficherSirius. - Exécutez votre programme avec
python dna.py databases/large.csv sequences/16.txt. Votre programme doit afficherNo match. - Exécutez votre programme avec
python dna.py databases/large.csv sequences/17.txt. Votre programme doit afficherHarry. - Exécutez votre programme avec
python dna.py databases/large.csv sequences/18.txt. Votre programme doit afficherNo match. - Exécutez votre programme avec
python dna.py databases/large.csv sequences/19.txt. Votre programme doit afficherFred. - Exécutez votre programme avec
python dna.py databases/large.csv sequences/20.txt. Votre programme doit afficherNo match.
Justesse
check50 cs50/problems/2024/x/dna
Style
style50 dna.py
Comment soumettre
submit50 cs50/problems/2024/x/dna